我院陆港院区是国家医学中心建设的重要承载地,在推进院区高质量内涵式发展进程中肩负重任。陆港儿科在承担大量儿童常见病、多发病医疗工作的同时,对于罕见病的诊治能力也不断提升。
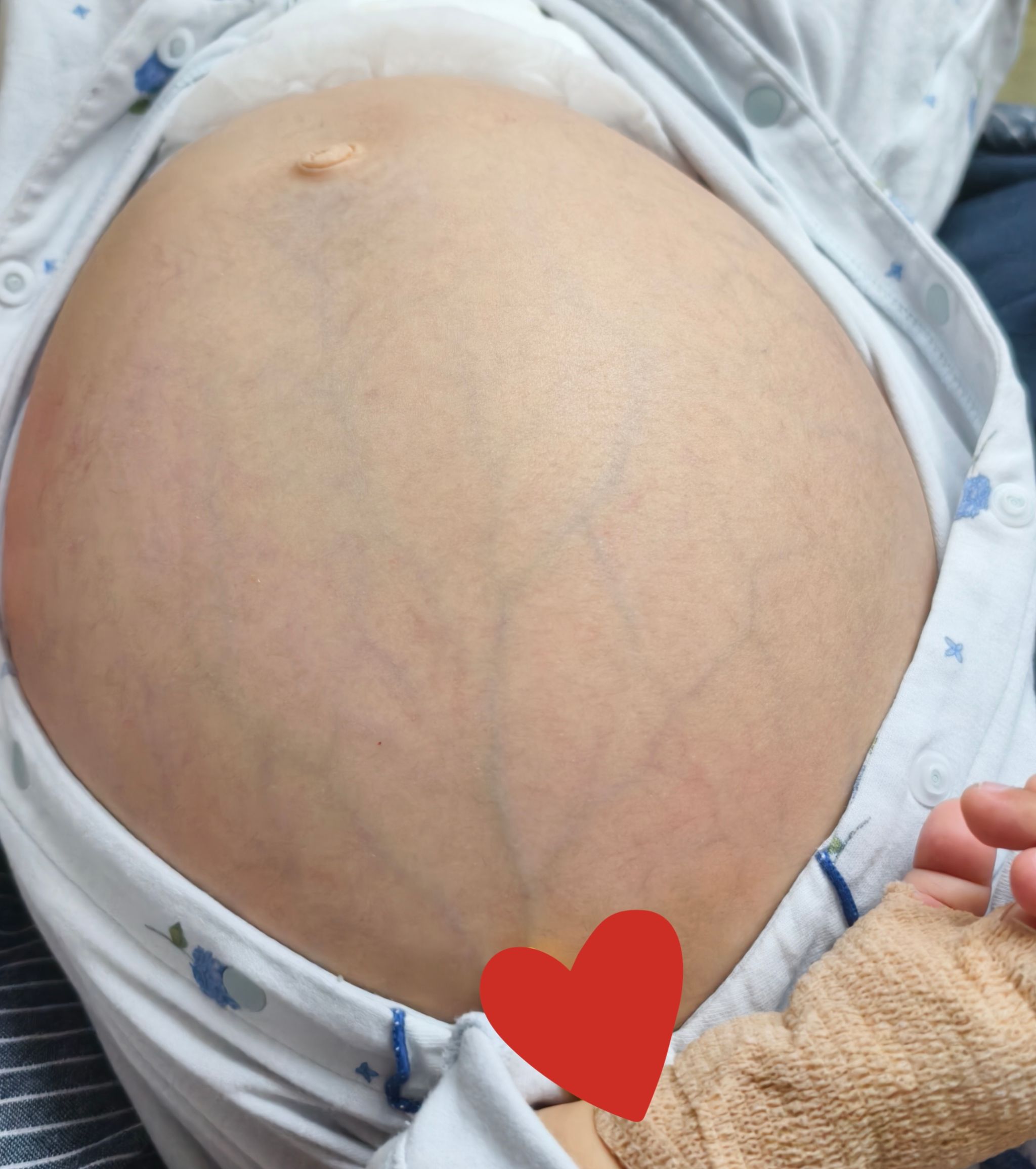
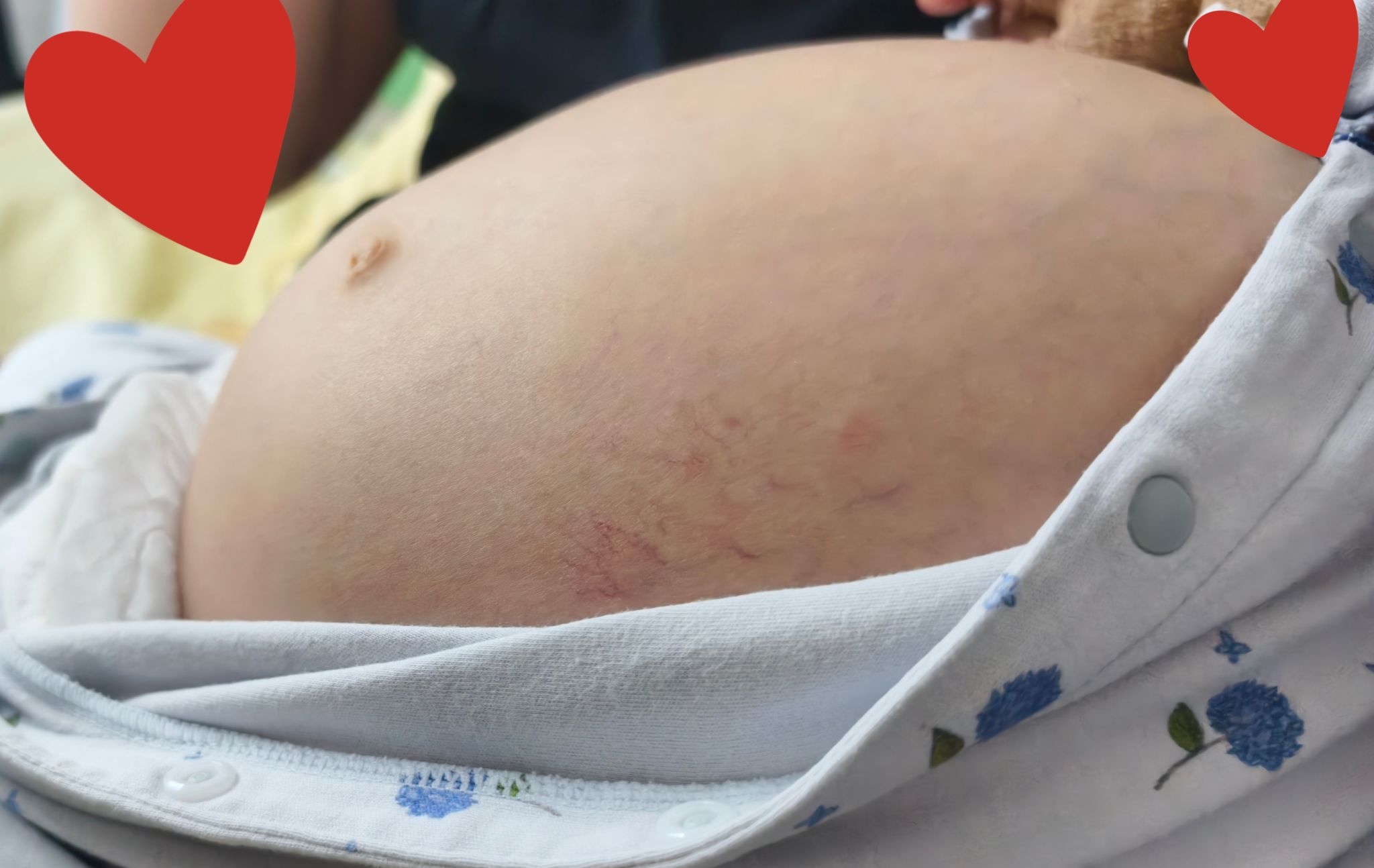
近日,我院陆港院区儿科收治了一名小幼儿,10月龄时妈妈发现孩子肚子变得越来越大,面色也发黄,外院检查发现原来是肝脾肿大的原因,还做了骨髓穿刺发现尼曼匹克细胞,基因检测结果提示SMPD1基因复合杂合变异,最终确诊为尼曼匹克病A型,该病是一种多系统受累的罕见疾病,且A型预后最差,最严重的一型,目前尚无特效治疗方案,该患儿口服药物治疗效果十分不理想。现在宝宝已经13月龄了,肚子鼓鼓的,近1周又出现了腹泻、呕吐,呕吐物夹杂少量血丝,肚皮越来越“硬”,小腿上还出现了瘀斑,转诊多家医院都建议转诊治疗。患儿家长多方打听得知我院儿科是陕西省罕见病专科联盟单位,满怀希望专程来到陆港院区儿科就诊。入院时患儿生命体征极不平稳,呼吸急促不能平卧,巩膜发黄,腹部高度膨隆,腹围达61厘米,腹壁静脉曲张明显,肝脾肿大达髂窝。杨永华副主任医师、何敏主治医师、贺鑫禹住院总医师等组建诊疗团队,快速评估病情,腹部超声显示患儿腹腔存在大量积液,伴重度肝脾肿大、脾静脉增宽。同时还合并低蛋白血症、全血细胞减少、凝血功能异常紊乱,患儿严重出血风险极高。
该患儿病情复杂且合并多系统损害,全科立刻启动紧急治疗措施,输血浆改善凝血功能、人血白蛋白及利尿以减轻腹水,同时还需要兼顾补液以维持水电解质内环境稳定,所以既要“利”还要“补”,这也是考验儿科医生基本功的一场实战。在家属的极力配合和儿科医护团队共同努力下,宝宝的肚子变“软”了,呼吸也平稳了,挺个小肚子坐在床上玩了起来。
尼曼-匹克病是一种常染色体隐性遗传病,临床病例罕见,分为三种亚型:A、B和C型。其中A型即酸性鞘磷脂酶缺乏症,是由于 SMPD1 基因突变所致,多于婴儿期起病,最早出现的症状是腹部膨隆,肝脾增大,智力和运动发育落后,多系统受累,多数在3岁左右死亡。目前缺乏有效的治愈方案,造血干细胞移植对于A、B 型患者可以在一定程度上缩小肝脾体积,延缓疾病进展,但对神经系统症状改善不明显。
此次凭借严密的病情观察、严谨的逻辑分析、果断的救治决策,患儿症状及相关实验室指标显著改善,转危为安,这一危重病人的收治及治疗的成功并非偶然,不仅标志着儿科全体医护人员用专业与坚守书写了生命护航的医者担当,也彰显了陆港院区儿科对罕见疾病的综合诊疗能力取得了重大突破,引领着陆港儿科病区迈向新的方向。未来我院陆港儿科将继续深耕疑难重症及罕见病的诊疗,不断提升团队诊疗质量,精进诊疗方案,用更精湛的技术与优质的服务,为陆港儿童的健康保驾护航。